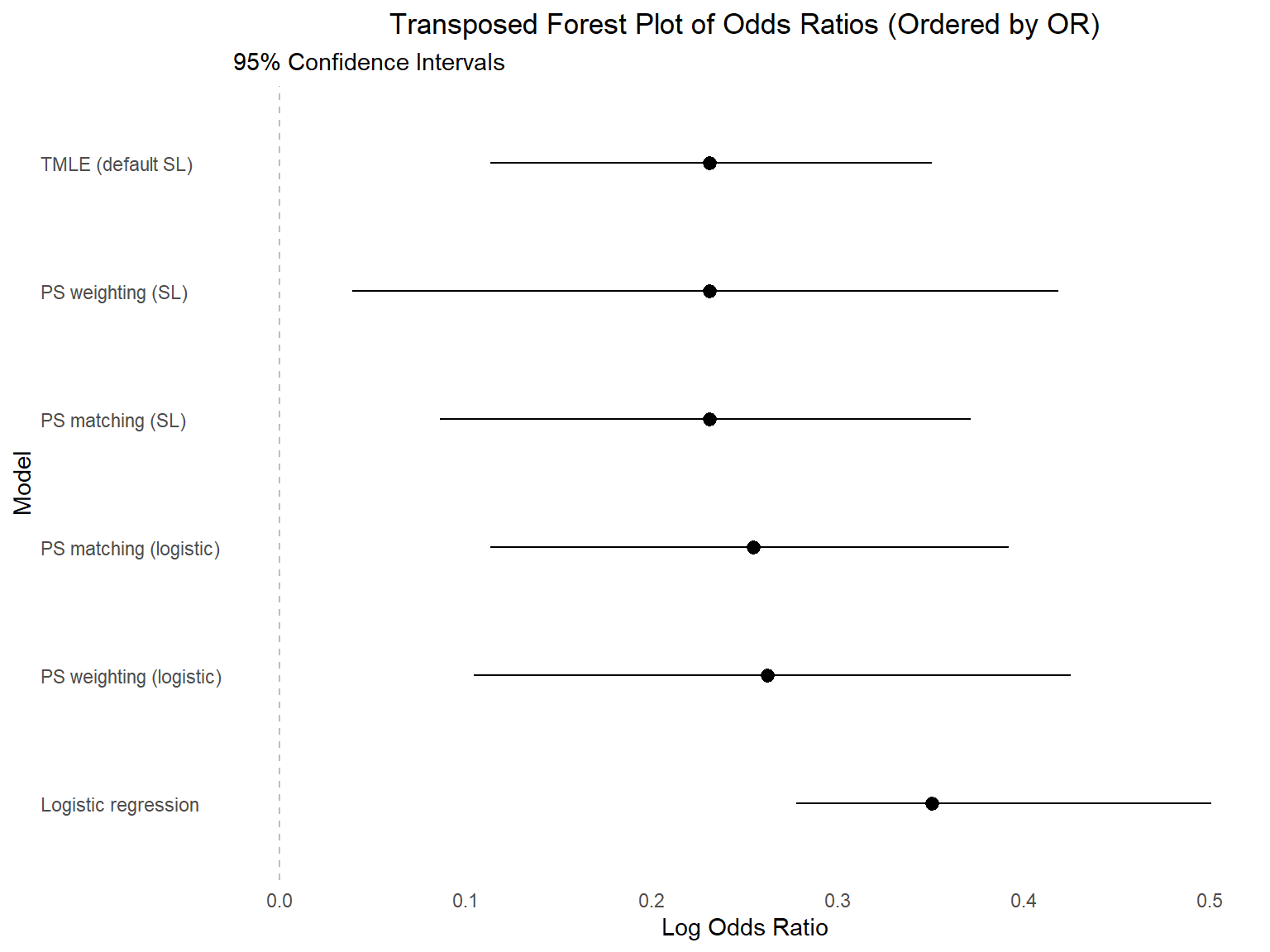
Comparing results
In this tutorial, we will use the RHC dataset in exploring the relationship between RHC (yes/no) and death (yes/no). We will use the following approaches:
- logistic regression
- propensity score matching and weighting with logistic regression
- propensity score matching and weighting with Super Learner
- TMLE
Load packages
We load several R packages required for fitting the models.
Load dataset
Confounder list in the baselinevars vector is
| Confounders |
|---|
| Disease.category |
| Cancer |
| Cardiovascular |
| Congestive.HF |
| Dementia |
| Psychiatric |
| Pulmonary |
| Renal |
| Hepatic |
| GI.Bleed |
| Tumor |
| Immunosupperssion |
| Transfer.hx |
| MI |
| age |
| sex |
| edu |
| DASIndex |
| APACHE.score |
| Glasgow.Coma.Score |
| blood.pressure |
| WBC |
| Heart.rate |
| Respiratory.rate |
| Temperature |
| PaO2vs.FIO2 |
| Albumin |
| Hematocrit |
| Bilirubin |
| Creatinine |
| Sodium |
| Potassium |
| PaCo2 |
| PH |
| Weight |
| DNR.status |
| Medical.insurance |
| Respiratory.Diag |
| Cardiovascular.Diag |
| Neurological.Diag |
| Gastrointestinal.Diag |
| Renal.Diag |
| Metabolic.Diag |
| Hematologic.Diag |
| Sepsis.Diag |
| Trauma.Diag |
| Orthopedic.Diag |
| race |
| income |
Logistic regression
Let us define the regression formula and fit logistic regression, adjusting for baseline confounders.
We can use flextable package to view fit.glm, the regression output:
Variable |
Odds_Ratio |
CI_Lower |
CI_Upper |
|---|---|---|---|
RHC.use |
1.42 |
1.23 |
1.65 |
As we can see, the odds of death was 42% higher among those participants with RHC use than those with no RHC use.
Propensity score matching with logistic regression
Now we will use propensity score matching, where propensity score will be estimated using logistic regression. The first step is to define the propensity score formula and estimate the propensity scores.
Step 1
# Propensity score model define
ps.formula <- as.formula(paste0("RHC.use ~ ",
paste(baselinevars,
collapse = "+")))
# Propensity score model fitting
fit.ps <- glm(ps.formula,
data = dat,
family = binomial)
# Propensity scores
dat$ps <- predict(fit.ps,
type = "response")
summary(dat$ps)
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 0.002478 0.161446 0.358300 0.380820 0.574319 0.968425Step 2
The second step is to match exposed (i.e., RHC users) to unexposed (i.e., RHC non-users) based on the propensity scores. We will use nearest neighborhood and caliper approach.
# See how many matched
match.obj
#> A `matchit` object
#> - method: 1:1 nearest neighbor matching without replacement
#> - distance: User-defined [caliper]
#> - caliper: <distance> (0.068)
#> - number of obs.: 5735 (original), 3478 (matched)
#> - target estimand: ATT
#> - covariates: too many to name
# Extract matched data
matched.data <- match.data(match.obj)
# Overlap checking
bal.plot(match.obj,
var.name="distance",
which="both",
type = "density",
colors = c("red","blue"))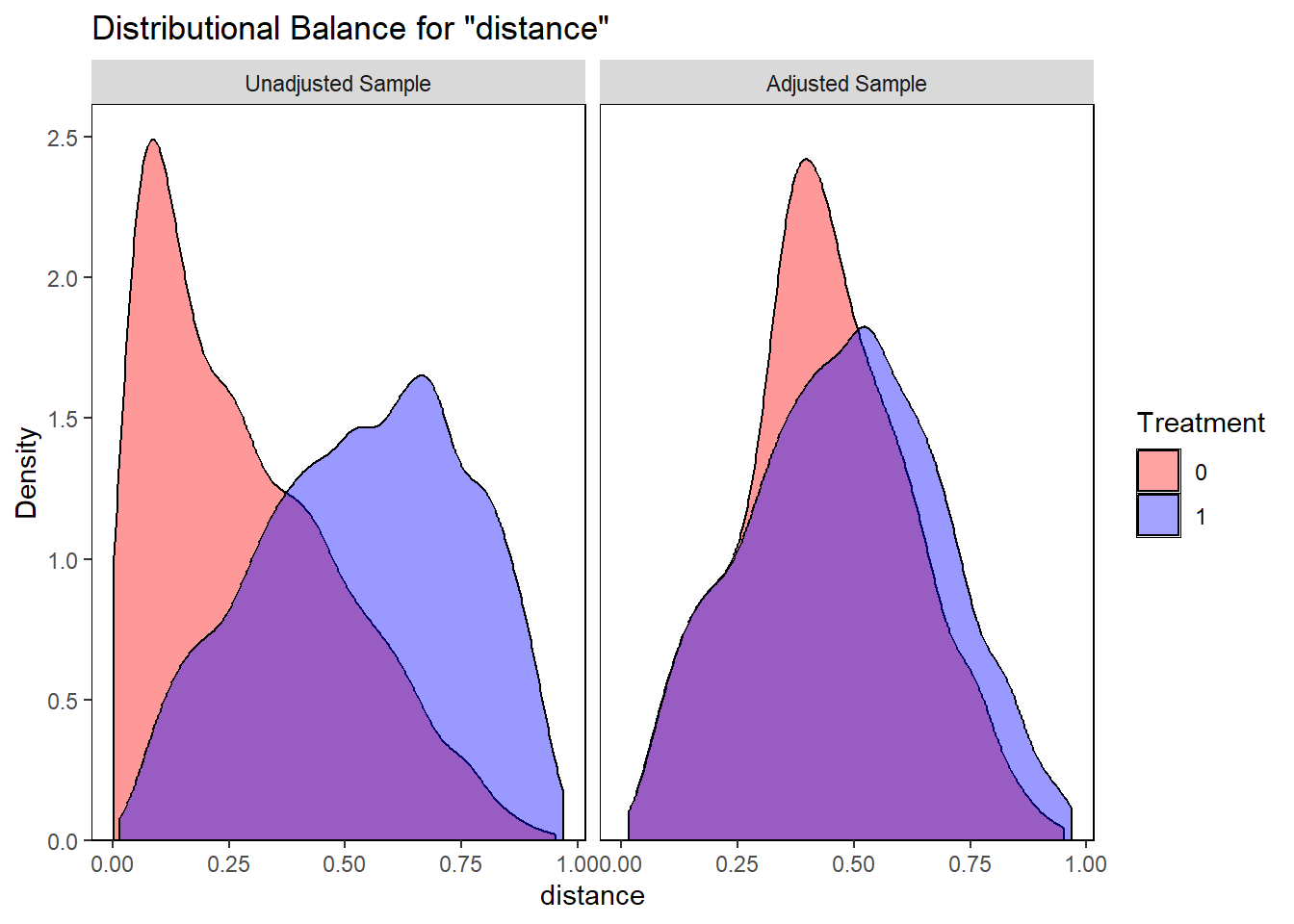
Step 3
The third step is to check the balancing on the matched data. We will compare the similarity of baseline characteristics between RHC users and non-users in the propensity score matched sample. Let’s consider SMD >0.1 as imbalanced.
# Balance checking in terms of SMD - using love plot
love.plot(match.obj,
binary = "std",
grid = TRUE,
thresholds = c(m = .1),
colors = c("red","blue")) 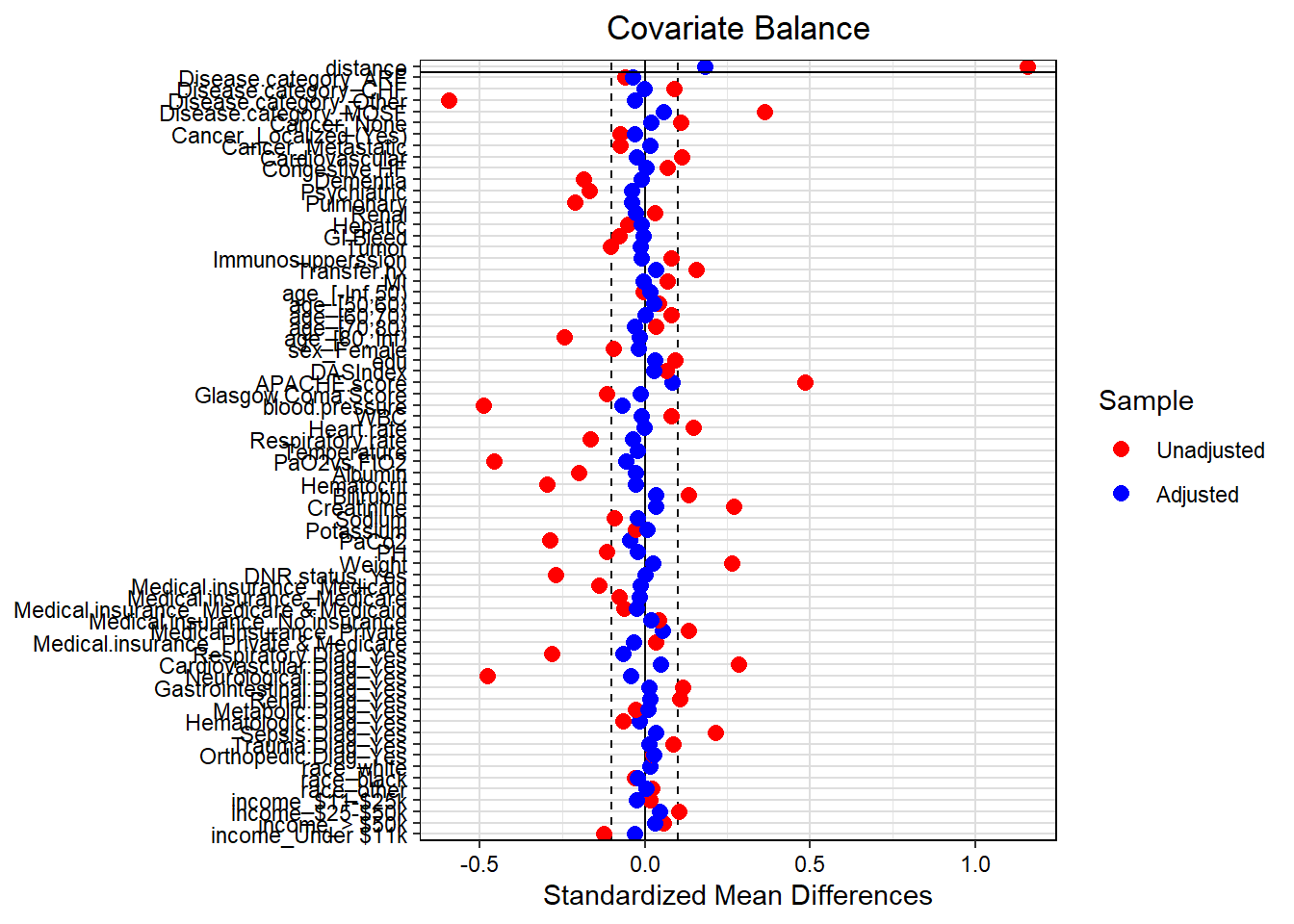
ExtractSmd(tab1b) shows
Variables |
1 vs 2 |
|---|---|
Disease.category |
0.06 |
Cancer |
0.03 |
Cardiovascular |
0.03 |
Congestive.HF |
0.00 |
Dementia |
0.01 |
Psychiatric |
0.04 |
Pulmonary |
0.04 |
Renal |
0.03 |
Hepatic |
0.01 |
GI.Bleed |
0.00 |
Tumor |
0.01 |
Immunosupperssion |
0.01 |
Transfer.hx |
0.03 |
MI |
0.01 |
age |
0.04 |
sex |
0.02 |
edu |
0.03 |
DASIndex |
0.03 |
APACHE.score |
0.08 |
Glasgow.Coma.Score |
0.01 |
blood.pressure |
0.07 |
WBC |
0.01 |
Heart.rate |
0.00 |
Respiratory.rate |
0.04 |
Temperature |
0.02 |
PaO2vs.FIO2 |
0.06 |
Albumin |
0.03 |
Hematocrit |
0.03 |
Bilirubin |
0.03 |
Creatinine |
0.03 |
Sodium |
0.02 |
Potassium |
0.01 |
PaCo2 |
0.04 |
PH |
0.02 |
Weight |
0.02 |
DNR.status |
0.00 |
Medical.insurance |
0.06 |
Respiratory.Diag |
0.06 |
Cardiovascular.Diag |
0.05 |
Neurological.Diag |
0.04 |
Gastrointestinal.Diag |
0.01 |
Renal.Diag |
0.02 |
Metabolic.Diag |
0.01 |
Hematologic.Diag |
0.01 |
Sepsis.Diag |
0.03 |
Trauma.Diag |
0.02 |
Orthopedic.Diag |
0.05 |
race |
0.02 |
income |
0.06 |
After propensity score matching, all the confounders are balanced in terms of SMD. Now, we will fit the outcome model on the matched data.
Step 4
Summary of fit.psm:
Variable |
Odds_Ratio |
CI_Lower |
CI_Upper |
|---|---|---|---|
RHC.use |
1.29 |
1.12 |
1.48 |
In the propensity score matched data, the odds of death was 29% higher among those participants with RHC use than those with no RHC use.
Propensity score weighting with logistic regression
Now we will use the propensity score weighting approach where propensity scores are estimated using logistic regression.
Step 1
Step 1 is the same as we did it for the propensity score matching.
Step 2
For the second step, we will calculate the stabilized inverse probability weight.
Step 3
Now, we will check the balance in terms of SMD.
# Design with inverse probability weights
w.design <- svydesign(id = ~1, weights = ~ipw, data = dat, nest = F)
# Balance checking in terms of SMD
tab1e <- svyCreateTableOne(vars = baselinevars, strata = "RHC.use",
data = w.design, includeNA = T,
addOverall = F, test = F)
#print(tab1e, showAllLevels = T, catDigits = 2, smd = T)ExtractSmd(tab1e) shows
Variables |
1 vs 2 |
|---|---|
Disease.category |
0.02 |
Cancer |
0.01 |
Cardiovascular |
0.01 |
Congestive.HF |
0.00 |
Dementia |
0.05 |
Psychiatric |
0.02 |
Pulmonary |
0.02 |
Renal |
0.01 |
Hepatic |
0.00 |
GI.Bleed |
0.02 |
Tumor |
0.00 |
Immunosupperssion |
0.01 |
Transfer.hx |
0.01 |
MI |
0.00 |
age |
0.05 |
sex |
0.03 |
edu |
0.00 |
DASIndex |
0.04 |
APACHE.score |
0.01 |
Glasgow.Coma.Score |
0.00 |
blood.pressure |
0.01 |
WBC |
0.04 |
Heart.rate |
0.02 |
Respiratory.rate |
0.00 |
Temperature |
0.01 |
PaO2vs.FIO2 |
0.00 |
Albumin |
0.03 |
Hematocrit |
0.02 |
Bilirubin |
0.01 |
Creatinine |
0.01 |
Sodium |
0.01 |
Potassium |
0.03 |
PaCo2 |
0.02 |
PH |
0.01 |
Weight |
0.02 |
DNR.status |
0.04 |
Medical.insurance |
0.03 |
Respiratory.Diag |
0.01 |
Cardiovascular.Diag |
0.01 |
Neurological.Diag |
0.01 |
Gastrointestinal.Diag |
0.01 |
Renal.Diag |
0.01 |
Metabolic.Diag |
0.00 |
Hematologic.Diag |
0.00 |
Sepsis.Diag |
0.01 |
Trauma.Diag |
0.01 |
Orthopedic.Diag |
0.01 |
race |
0.02 |
income |
0.02 |
All confounders are balanced (SMD < 0.1).
Step 4
Summary of fit.ipw:
Variable |
Odds_Ratio |
CI_Lower |
CI_Upper |
|---|---|---|---|
RHC.use |
1.30 |
1.11 |
1.53 |
In the propensity score weighted data, the odds of death was 30% higher among those participants with RHC use than those with no RHC use.
Propensity score matching with super learner
Now we will use the propensity score matching, where we will be using super learner to calculate the propensity scores. We use logistic, LASSO, and XGBoost as the candidate learners.
Step 1
# Propensity scores for all learners
predictions <- cbind(ps.fit$SL.predict, ps.fit$library.predict)
head(predictions)
#> SL.glm_All SL.glmnet_All SL.xgboost_All
#> [1,] 0.3632635 0.39789364 0.39569431 0.32144672
#> [2,] 0.5870492 0.64983590 0.61497078 0.54465985
#> [3,] 0.5740246 0.66329951 0.65396650 0.46847290
#> [4,] 0.2172501 0.33044244 0.34478278 0.02363573
#> [5,] 0.6347749 0.45972157 0.43779434 0.86645132
#> [6,] 0.0272532 0.03420477 0.03629319 0.01730520
# Propensity scores for super learner
dat$ps.sl <- predictions[,1]
summary(dat$ps.sl)
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 0.001671 0.147762 0.337664 0.376282 0.587237 0.975584Step 2
The same as before, we will match exposed to unexposed based on their propensity scores.
# See how many matched
match.obj2
#> A `matchit` object
#> - method: 1:1 nearest neighbor matching without replacement
#> - distance: User-defined [caliper]
#> - caliper: <distance> (0.075)
#> - number of obs.: 5735 (original), 3430 (matched)
#> - target estimand: ATT
#> - covariates: too many to name
# Extract matched data
matched.data.sl <- match.data(match.obj2)
# Overlap checking
bal.plot(match.obj2,
var.name="distance",
which="both",
type = "density",
colors = c("red","blue"))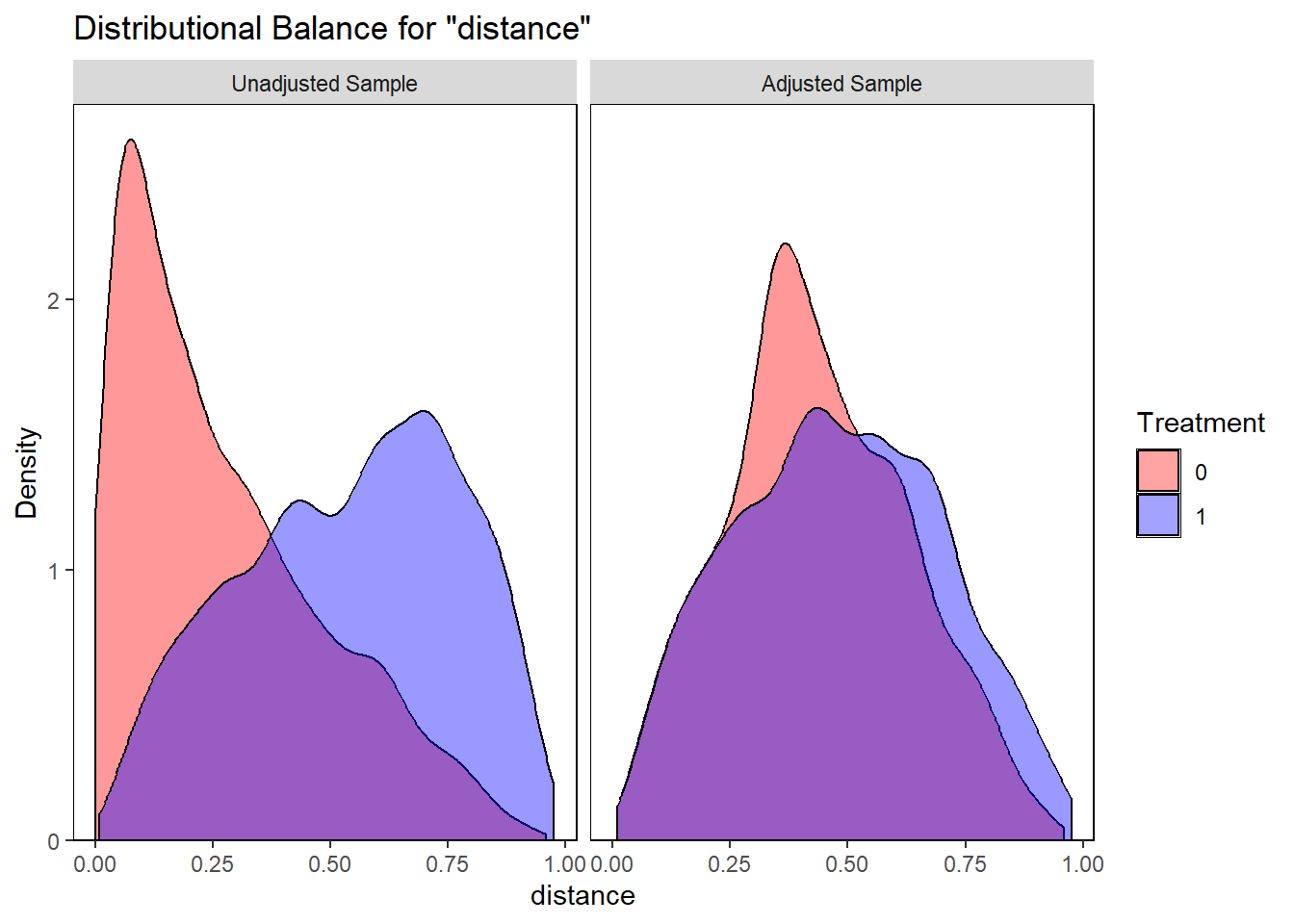
Step 3
Now we will check the balancing on the matched data.
# Balance checking in terms of SMD - using love plot
love.plot(match.obj2,
binary = "std",
grid = TRUE,
thresholds = c(m = .1),
colors = c("red","blue")) 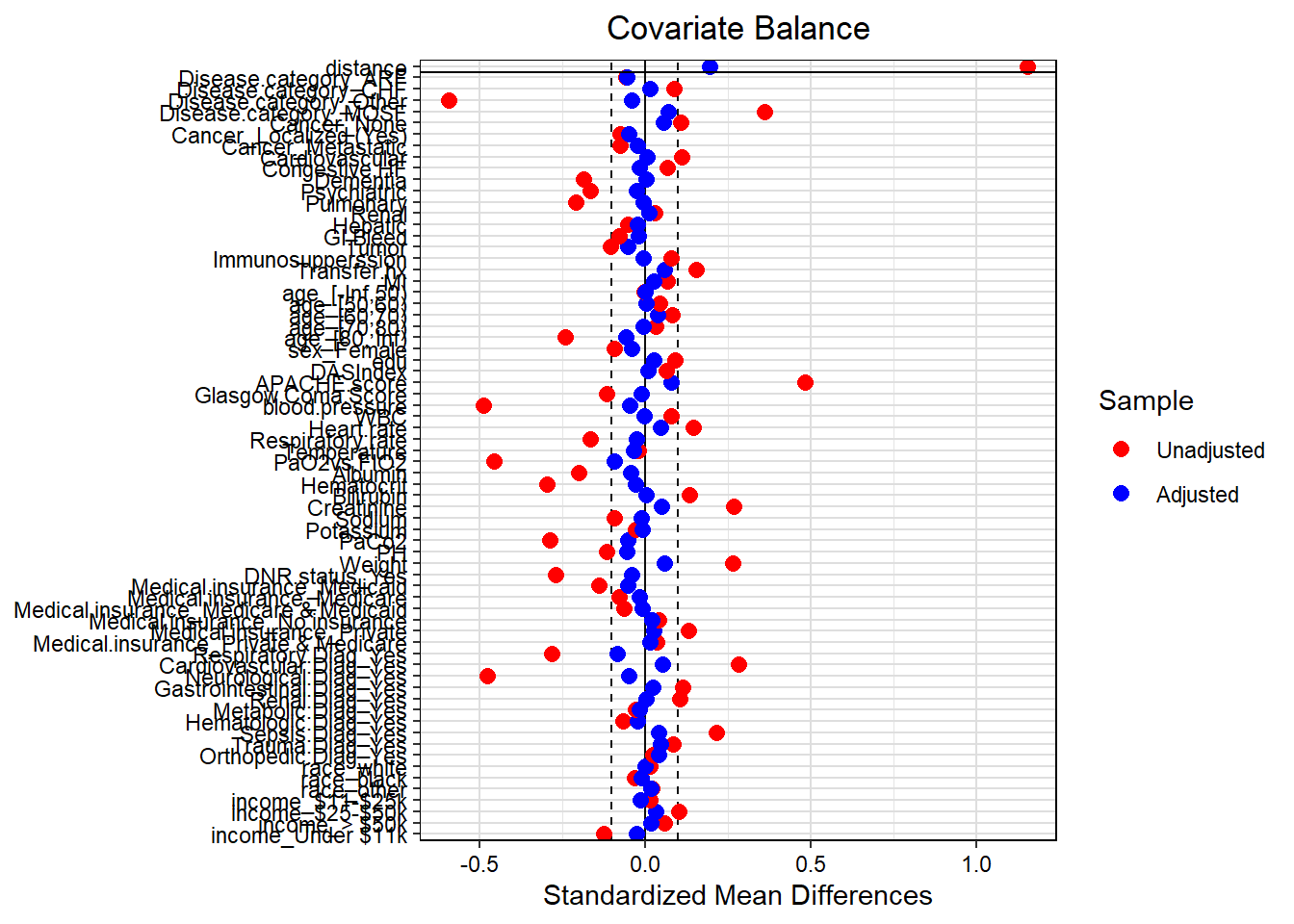
ExtractSmd(tab1c) shows
Variables |
1 vs 2 |
|---|---|
Disease.category |
0.08 |
Cancer |
0.05 |
Cardiovascular |
0.01 |
Congestive.HF |
0.02 |
Dementia |
0.00 |
Psychiatric |
0.02 |
Pulmonary |
0.01 |
Renal |
0.01 |
Hepatic |
0.02 |
GI.Bleed |
0.02 |
Tumor |
0.05 |
Immunosupperssion |
0.01 |
Transfer.hx |
0.06 |
MI |
0.03 |
age |
0.06 |
sex |
0.04 |
edu |
0.03 |
DASIndex |
0.01 |
APACHE.score |
0.08 |
Glasgow.Coma.Score |
0.01 |
blood.pressure |
0.05 |
WBC |
0.00 |
Heart.rate |
0.05 |
Respiratory.rate |
0.03 |
Temperature |
0.04 |
PaO2vs.FIO2 |
0.09 |
Albumin |
0.05 |
Hematocrit |
0.03 |
Bilirubin |
0.00 |
Creatinine |
0.05 |
Sodium |
0.01 |
Potassium |
0.01 |
PaCo2 |
0.05 |
PH |
0.06 |
Weight |
0.06 |
DNR.status |
0.04 |
Medical.insurance |
0.06 |
Respiratory.Diag |
0.08 |
Cardiovascular.Diag |
0.05 |
Neurological.Diag |
0.04 |
Gastrointestinal.Diag |
0.02 |
Renal.Diag |
0.00 |
Metabolic.Diag |
0.02 |
Hematologic.Diag |
0.02 |
Sepsis.Diag |
0.04 |
Trauma.Diag |
0.06 |
Orthopedic.Diag |
0.06 |
race |
0.02 |
income |
0.04 |
Again, all confounders are balanced in terms of SMD (all SMDs < 0.1). Next, we will fit the outcome model.
Step 4
Summary of fit.psm.sl:
Variable |
Odds_Ratio |
CI_Lower |
CI_Upper |
|---|---|---|---|
RHC.use |
1.26 |
1.09 |
1.45 |
The interpretation is the same as before. In the propensity score matched data, the odds of death was 26% higher among those participants with RHC use than those without RHC use.
Propensity score weighting with super learner
Step 1
Step 1 is the same as we did it for the propensity score matching.
Step 2
For the second step, we will calculate the stabilized inverse probability weight.
Step 3
Next, we will check the balance in terms of SMD.
# Design with inverse probability weights
w.design.sl <- svydesign(id = ~1, weights = ~ipw.sl,
data = dat, nest = F)
# Balance checking in terms of SMD
tab1k <- svyCreateTableOne(vars = baselinevars, strata = "RHC.use",
data = w.design.sl, includeNA = T,
addOverall = T, test = F)
#print(tab1k, showAllLevels = T, catDigits = 2, smd = T)ExtractSmd(tab1k) shows
Variables |
1 vs 2 |
|---|---|
Disease.category |
0.01 |
Cancer |
0.03 |
Cardiovascular |
0.02 |
Congestive.HF |
0.01 |
Dementia |
0.05 |
Psychiatric |
0.03 |
Pulmonary |
0.01 |
Renal |
0.01 |
Hepatic |
0.00 |
GI.Bleed |
0.02 |
Tumor |
0.00 |
Immunosupperssion |
0.01 |
Transfer.hx |
0.00 |
MI |
0.01 |
age |
0.07 |
sex |
0.03 |
edu |
0.00 |
DASIndex |
0.04 |
APACHE.score |
0.01 |
Glasgow.Coma.Score |
0.02 |
blood.pressure |
0.05 |
WBC |
0.06 |
Heart.rate |
0.00 |
Respiratory.rate |
0.01 |
Temperature |
0.00 |
PaO2vs.FIO2 |
0.03 |
Albumin |
0.00 |
Hematocrit |
0.02 |
Bilirubin |
0.01 |
Creatinine |
0.00 |
Sodium |
0.00 |
Potassium |
0.02 |
PaCo2 |
0.05 |
PH |
0.02 |
Weight |
0.01 |
DNR.status |
0.05 |
Medical.insurance |
0.03 |
Respiratory.Diag |
0.02 |
Cardiovascular.Diag |
0.01 |
Neurological.Diag |
0.02 |
Gastrointestinal.Diag |
0.00 |
Renal.Diag |
0.01 |
Metabolic.Diag |
0.01 |
Hematologic.Diag |
0.01 |
Sepsis.Diag |
0.00 |
Trauma.Diag |
0.05 |
Orthopedic.Diag |
0.02 |
race |
0.05 |
income |
0.05 |
All confounders are balanced (SMD < 0.1).
Step 4
Summary of fit.ipw.sl:
Variable |
Odds_Ratio |
CI_Lower |
CI_Upper |
|---|---|---|---|
RHC.use |
1.26 |
1.04 |
1.52 |
In the propensity score weighted data, the odds of death was 26% higher among those participants with RHC use than those without RHC use.
TMLE
From the previous tutorial, we calculated the effective sample size for the outcome model as well as for the exposure model:
Since the effective sample size is \(\ge 5,000\) for both model, we can consider \(5 \le \text{V} \le 10\), where V is the number of folds. Let’s work with 10-fold cross-validation for both the exposure and the outcome model, with the default super learner library.
As we can see that the odds of death was 26% higher among those participants with RHC use than those without RHC use.
Results comparison
| Model | OR | 95% CI |
|---|---|---|
| Logistic regression | 1.42 | 1.32-1.65 |
| Propensity score matching with logistic | 1.29 | 1.12-1.48 |
| Propensity score weighting with logistic | 1.30 | 1.11-1.53 |
| Propensity score matching with super learner (logistic, LASSO, and XGBoost) | 1.26 | 1.09-1.45 |
| Propensity score weighting with super learner (logistic, LASSO, and XGBoost) | 1.26 | 1.04-1.52 |
| TMLE (default SL library) | 1.26 | 1.12-1.42 |